OPEN-ACCESS PEER-REVIEWED
RESEARCH ARTICLE
Sjoukje Postma-Kunnen1,†, Jan Peter Yska1,†,*, Gerard Hommema1, Sikke Koopmans1, Bob Wilffert2,3, Eric N. van Roon1,2
1Department of Clinical Pharmacy and Clinical Pharmacology, Medical Centre Leeuwarden, Leeuwarden, the Netherlands. 2University of Groningen, Groningen Research Institute of Pharmacy, PharmacoTherapy, -Epidemiology & -Economics, Groningen, the Netherlands. 3Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen, University of Groningen, Groningen, the Netherlands.†Both authors contributed equally to this work.
Journal of Applied Bioanalysis. Vol.3. No.3. pages 49-57 (2017)
Published 15 April 2017. https://doi.org/10.17145/jab.17.008 | (ISSN 2405-710X)
*Correspondence:
Yska JP . Department of Clinical Pharmacy and Clinical Pharmacology, Medical Centre Leeuwarden, POBox 888, 8901 BR Leeuwarden, The Netherlands. Phone: +31 582866604; Fax: +31 582866606
Citation:
Postma-Kunnen S, Yska JP, Gerard Hommema G, Koopmans S, Wilffert B, van Roon EN. A validated high-resolution accurate mass LC-MS assay for quantitative determination of metoprolol and α-hydroxymetoprolol in human serum for application in pharmacokinetics. J Appl Bioanal 3(3), 49-57 (2017).
Open-access and Copyright:
©2017 Postma-Kunnen et al. This article is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Funding/Manuscript writing assistance:
The authors have no financial support or funding to report and authors declare that no writing assistance was utilized in the production of this article.
Competing interest:
The authors have declared that no competing interest exist.
Article history:
Received: 20 March 2017, Revised 10 May 2017, Accepted 14 May 2017.
Abstract
To determine metoprolol and its metabolite α-hydroxymetoprolol in human serum we validated a method on an LC system with an Exactive® Orbitrap mass spectrometer (Thermo Scientific) as detector and isotope-labelled metoprolol-d7 as internal standard. A simple sample preparation was used with water-acetonitrile (15:85, v/v) as precipitation reagent. This method has a chromatographic run time of 15 min and linear calibration curves in the range of 5.0-250 µg/L for both metoprolol and α-hydroxymetoprolol. Validation showed the method to be accurate, with a good precision, selective and with a lower limit of quantitation of 2.0 µg/L for metoprolol and 1.0 µg/L for α-hydroxymetoprolol, respectively. This validated LC-Orbitrap MS analysis for metoprolol and α-hydroxymetoprolol can be used for application in human pharmacokinetics.
Keywords
Metoprolol, α-hydroxymetoprolol, LC-MS, Orbitrap mass spectrometer, pharmacokinetics.
Introduction
Metoprolol, a lipophilic cardioselective β1-adrenoreceptor antagonist, is widely used in the treatment of hypertension, angina pectoris and other cardiovascular diseases. For oral administration it is available as immediate release (metoprolol tartrate) or controlled release tablet (metoprolol tartrate, metoprolol succinate). Metoprolol is almost completely absorbed from the gastrointestinal tract. However, due to an extensive first pass effect, only 50% of the dose reaches the systemic circulation. In the liver metoprolol is primarily metabolized by cytochrome P450 2D6 (CYP2D6) into several metabolites. α-hydroxymetoprolol (Figure 1) is an active metabolite but possesses only around one tenth of the β1-blocking activity of metoprolol. Serum concentrations of α-hydroxymetoprolol vary depending on age and on the oxidation phenotype [1].
To determine metoprolol and its metabolite α-hydroxymetoprolol in serum for application in pharmacokinetics we needed a convenient and accurate method of analysis. Numerous methods of analysis have been reported for determination of metoprolol, with and without metabolites, in human plasma, serum or urine, including high performance liquid chromatography with UV or fluorescence detection [2-7] and gas chromatography with mass spectrometry [8]. In recent years several methods using liquid chromatography with tandem mass spectrometry have been published [9-15]. So far, liquid chromatography with high-resolution accurate mass spectrometry (MS) using Orbitrap technology has rarely been used for application in therapeutic drug monitoring and pharmacokinetics. The aim of the present study was to validate a simple, sensitive LC–MS method to quantify metoprolol and α-hydroxymetoprolol in serum for application in human pharmacokinetics.
Materials and methods
Chemicals and reagents
Metoprolol tartrate, of pharmaceutical quality (Ph Eur), was procured by Astra Zeneca (Zoetermeer, The Netherlands) and Fagron (Capelle aan den IJssel, The Netherlands). Metoprolol 1.0 mg/mL in methanol, α-hydroxymetoprolol 100 mg/L in methanol, and metoprolol-d7 hydrochloride 100 mg/L in methanol were obtained from Campro Scientific (Veenendaal, The Netherlands). Methanol HPLC, gradient grade, and acetonitrile HPLC, gradient grade, were purchased from Boom BV (Meppel, The Netherlands). Methanol Lichrosolv and formic acid were obtained from Merck KGaA (Darmstadt, Germany). Ammonium formate was obtained from Acros Organics (Geel, Belgium). All chemicals were of suitable analytical grade. Newborn calf serum was purchased from Gibco (Thermo Fisher, Life Technologies Europe BV, Bleiswijk, The Netherlands). Blank human serum was obtained from unpaid, healthy volunteers. Purified water (sterile water for irrigation) was from Baxter (Utrecht, The Netherlands).
Liquid chromatography and mass spectrometric conditions
The liquid chromatography system (Thermo Scientific Dionex Ultimate 3000, Waltham, MA, USA) consisted of a binary Ultimate 3000 DGP-3600 RS pump, an online solvent degasser, a temperature-controlled Ultimate 3000 column compartment, and an Ultimate 3000 autosampler. Chromatographic separation was performed on a Thermo Scientific Hypersil Gold PFP 3 μm, 150 x 2.1 mm column. The column was maintained at 30 °C. The flow rate of the mobile phase was kept at 0.4 mL/min. The mobile phase consisted of (A) 2 mM ammonium formate and 0.1% (v/v) formic acid in water and (B) 0.1% (v/v) formic acid in methanol. The gradient elution method was as follows: 5% B at 0-0.5min, 5-95% B at 0.5-9.0 min, 95% B at 9.0-9.5 min, 95-5% B at 9.5-9.6 min, and 5% B at 9.6-15 min.
The autosampler temperature was maintained at 10 °C and the pressure of the system was 2600 psi. Detection of the analytes and the internal standard was performed on a high-resolution accurate mass Exactive® mass spectrometer equipped with an Orbitrap mass analyzer (Thermo Scientific, Waltham, MA, USA). The Orbitrap mass analyzer is a mass analyzer that traps ions in an orbital motion. The image current from the trapped ions is detected and converted into a mass spectrum using the Fourier transform of the frequency signal. Source dependent parameters for H-ESI II probe were: sheath gas pressure: 50 psi; auxiliary gas pressure: 30 psi; ion spray voltage: 3000 V; capillary temperature: 350 °C; vaporizer temperature: 300 °C. The scan parameters for the Exactive mass spectrometer were: full mass spectrum; microscan:1; resolution: 100,000; target AGC value: 1e6; maximum IT: 250 msec; scan range: 100-1000 m/z. MS acquisition method: PRM. Processing method parameters: metoprolol m/z 268.1907 mass tolerance ± 10 ppm; α-hydroxymetoprolol m/z 284.1856 mass tolerance ± 10 ppm; metoprolol-d7 m/z 275.2335 mass tolerance ± 10 ppm. Xcalibur software version 1.1 SP3 (Thermo Scientific) was used for instrument control, data m/z 284.1856 mass tolerance ± 10 ppm; metoprolol-d7 m/z 275.2335 mass tolerance ± 10 ppm. Xcalibur software version 1.1 SP3 (Thermo Scientific) was used for instrument control, data acquisition and processing.
Stock solutions, calibration standards and quality control samples
Stock solutions of metoprolol (5 mg/L) and α-hydroxymetoprolol (5 mg/L) were prepared in methanol. From both stock solutions 0,5 mL was diluted to 10.0 mL with blank calf serum to yield a working solution with a concentration of 250 µg/L for both metoprolol and α-hydroxymetoprolol. This solution was used for daily preparation of calibration standards at concentrations of 5.0, 10.0, 30.0, 60.0, 125.0, 185.0, 250.0 µg/L for both metoprolol and α-hydroxymetoprolol by diluting with blank calf serum. The quality control samples were prepared in blank calf serum at four different concentration levels: 5.0, 10.0 (LQ), 125.0 (MQ), 250.0 µg/L (HQ) for both metoprolol and α-hydroxymetoprolol respectively. For determination of the lower limit of quantitation (LLOQ) quality control samples of 0.5, 1.0, 2.0, 3.0, 4.0 and 5.0 µg/L for both metoprolol and α-hydroxymetoprolol were prepared by diluting the working standard solution with blank calf serum. Stock solution (10 mg/L) of the internal standard (IS) was prepared in a 10 mL volumetric flask by dilution of 1.00 mL of metoprolol-d7 hydrochloride (100 mg/L in methanol) with methanol. Precipitation solution with internal standard (0.025mg/L) was prepared in a 200 ml volumetric flask by dilution of 500 µL of stock solution of metoprolol-d7 hydrochloride (10 mg/L) with water:acetonitrile (15-85, v/v). All the solutions were stored at 2-8 °C and were brought to room temperature before use.
Sample preparation
An aliquot of 200 µL of serum sample was transferred into a 5 mL borosilicate glass test tube, and 750 µL of precipitation solution with IS was added. The mixture was vortexed for 5 sec. Next, the test tube was shaken on a Multi Reax shaker (Heidolph Instruments, Schwabach, Germany) for 5 min at 1400 rpm. The sample was then centrifuged for 5 min at 5000 rpm. An aliquot of 250 µL of the clear upper layer was transferred into a clean 10 mL borosilicate glass test tube. The sample was evaporated to dryness in an IR-dancer (Hettlab AG, Bäch, Switzerland) for 30 min at 30-35 °C. The residue was reconstituted with 250 µL of the mobile phase (A-B 95:5 (v/v)). The sample was transferred into a pre-labeled autosampler vial and 10 µl was used for injection into the chromatographic system.
Bioanalytical method validation
Method validation
The method was validated largely in accordance with the EMEA Guideline on bioanalytical method validation [16]. The validated criteria included selectivity, linearity, accuracy and precision, matrix effects and stability. The validation was performed with maximum tolerated bias and relative standard deviation (RSD) of 20% for the lower limit of quantitation and 15% for the other validated concentrations.
Matrix effects
Blank calf serum was used as surrogate matrix for making calibrators and quality control samples. For comparison of the effect of surrogate matrix calf serum vs human serum on recovery, at each level of calibration blank human serum samples were spiked with metoprolol and α-hydroxymetoprolol. Samples were analyzed on a calibration curve obtained from blank calf serum calibrators. Mean recoveries should be within 85-115%. F-test and T-test should show no significant difference on recovery (p<0.05) between surrogate matrix blank calf serum and blank human serum.
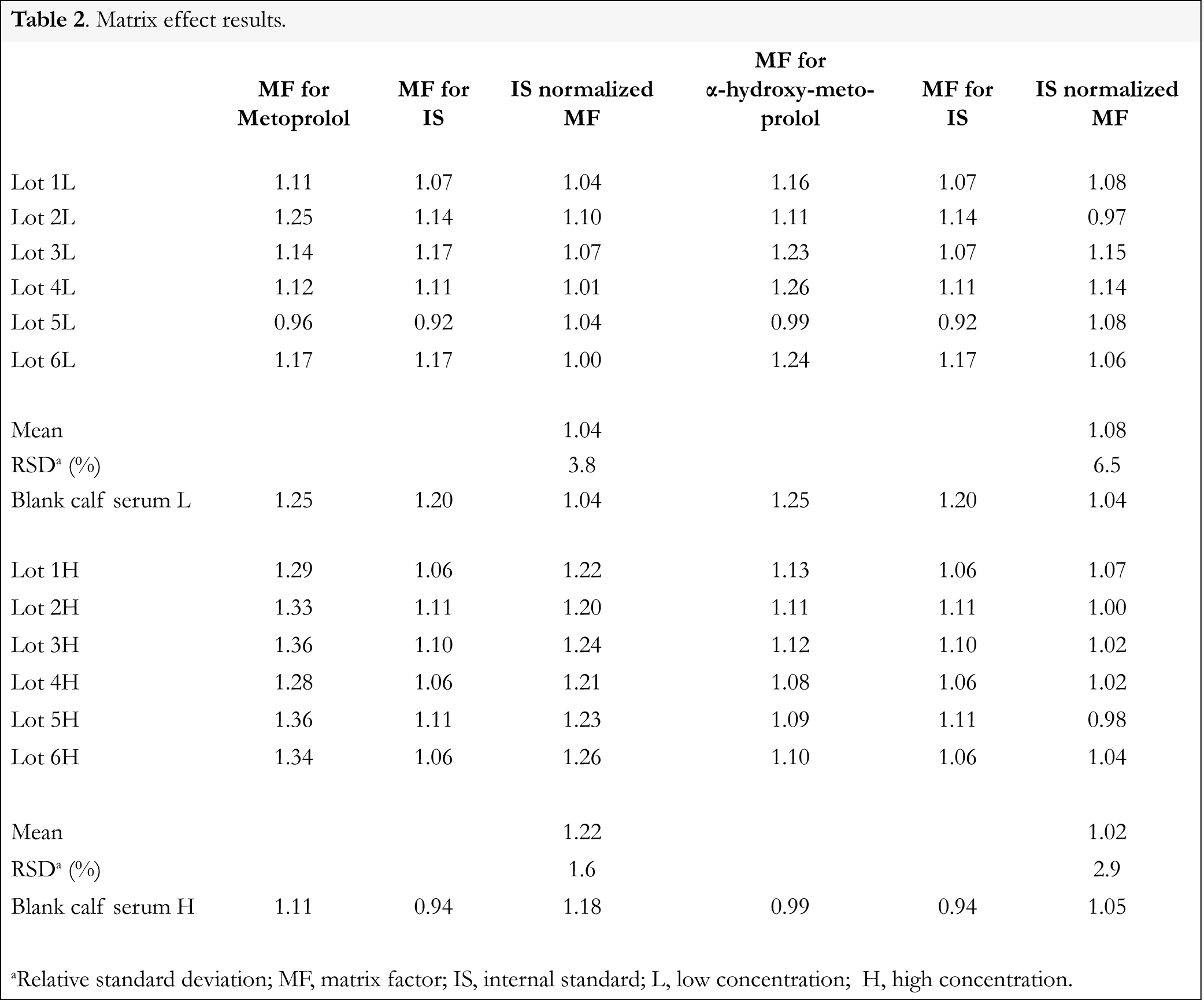
For the evaluation of the effects of human serum constituents over the ionization of metoprolol, α-hydroxymetoprolol and IS 6 different lots of blank human serum from individual donors were used. For metoprolol, α-hydroxymetoprolol and IS the matrix factor (MF) was calculated for each lot of human serum by calculating the ratio of the peak area in the presence of matrix (measured by analyzing blank human serum spiked after extraction with analyte), to the peak area in absence of matrix (precipitation solution spiked with analyte). The IS normalized MF was also calculated by dividing the MF of the analyte by the MF of the IS. The relative standard deviation (RSD) of the IS normalized MF calculated from the 6 lots of blank human serum should not be greater than 15% [10]. Matrix effect for metoprolol and α-hydroxymetoprolol were determined at low and high concentrations i.e., 10.0 and 250.0 µg/L, whereas the matrix effect over the IS was determined at a single concentration of 25.0 µg/L.
Selectivity
The selectivity of the method was determined by analysis of blank calf serum, blank human serum and serum from 6 different patients. Every sample was processed by the procedure as described in section 2.4. and injected into the chromatographic system in order to determine the extent of interference of endogenous serum components at the retention times of metoprolol, α-hydroxymetoprolol and IS.
Linearity and lower limit of quantitation
To evaluate linearity, calibration standards of 7 concentrations of metoprolol (5-250 µg/L) and α-hydroxymetoprolol (5-250 µg/L) were separately extracted and assayed. Three calibration curves were determined at 3 separate days by different analysts. The linearity of each calibration curve of metoprolol and α-hydroxymetoprolol was determined by plotting the peak area ratios of the analyte and IS peaks against concentrations. The calibration model was selected based on the analysis of the data by linear regression with weighting factors (none, 1/x, and 1/x2). The quantitation limit is generally determined by the analysis of samples with known concentrations of analyte and by establishing the minimum level at which the analyte can be quantified with acceptable accuracy and precision. For determination of LLOQ, 6 replicates of quality control samples of metoprolol and α-hydroxymetoprolol in concentrations ranging from 0.5 to 5.0 µg/L were analyzed against the calibration curve at 3 separate days and the obtained concentrations were compared with the nominal value. The mean within-run and between-run concentrations of the LLOQ should be within 20% of the nominal value. The within-run and between run relative standard deviation (RSD, %) should not exceed 20%.
Precision and accuracy
The precision and accuracy of the method were evaluated by assaying QC samples at 5 concentrations with 6 replicates for each concentration. Six replicates of each level were analyzed in a day or over 3 separate days by different analysts by performing the repeatability of the analysis. Precision was expressed as relative standard deviation (RSD, %), and accuracy was expressed as the relative error (RE, %) from the nominal value. The deviation in precision value (RSD, %) should be within 15% and accuracy (RE, %) should be within 15% from the nominal values, except for LLOQ, where it should not exceed 20%.
Stability
Stability experiments were performed to evaluate the stability of metoprolol and α-hydroxymetoprolol during sample preparation and sample analysis. To determine bench top stability 6 replicates of each level of LQ and HQ samples were maintained at room temperature in daylight or in the dark for 24 h. Then the samples were processed and analyzed. Long term stability with storage at -24 °C for 0.75 y was determined at LQ, MQ and HQ levels for 2 replicates of each level. Then the samples were processed and analyzed. Freeze-thaw stability with storage at -24 °C was performed at LQ, MQ and HQ levels using 6 replicates at each level. After the completion of 2 freeze-thaw cycles the samples were analyzed. Autosampler stability was performed at LQ and HQ levels using 16 replicates at each level and processing 1 sample every 2 h for 30 h. For all experiments the concentrations of metoprolol and α-hydroxymetoprolol were compared with the nominal values. Mean recovery and recovery in comparison with baseline were determined. The mean recovery at each level should be within ±15% of the nominal concentration.
Results and discussion
Method development
For use in clinical toxicology in our laboratory an ultra-high resolution accurate mass LC/MS assay for forensic toxicology screening in human serum was slightly modified [17]. The time of the gradient elution method was reduced to 15 min and sample preparation was performed with water-acetonitril 15:85, v/v. This clinical toxicology screening method was used for quantitative determination of metoprolol and α-hydroxymetoprolol.
Figures and Tables



[Click to enlarge]
Separation and detection
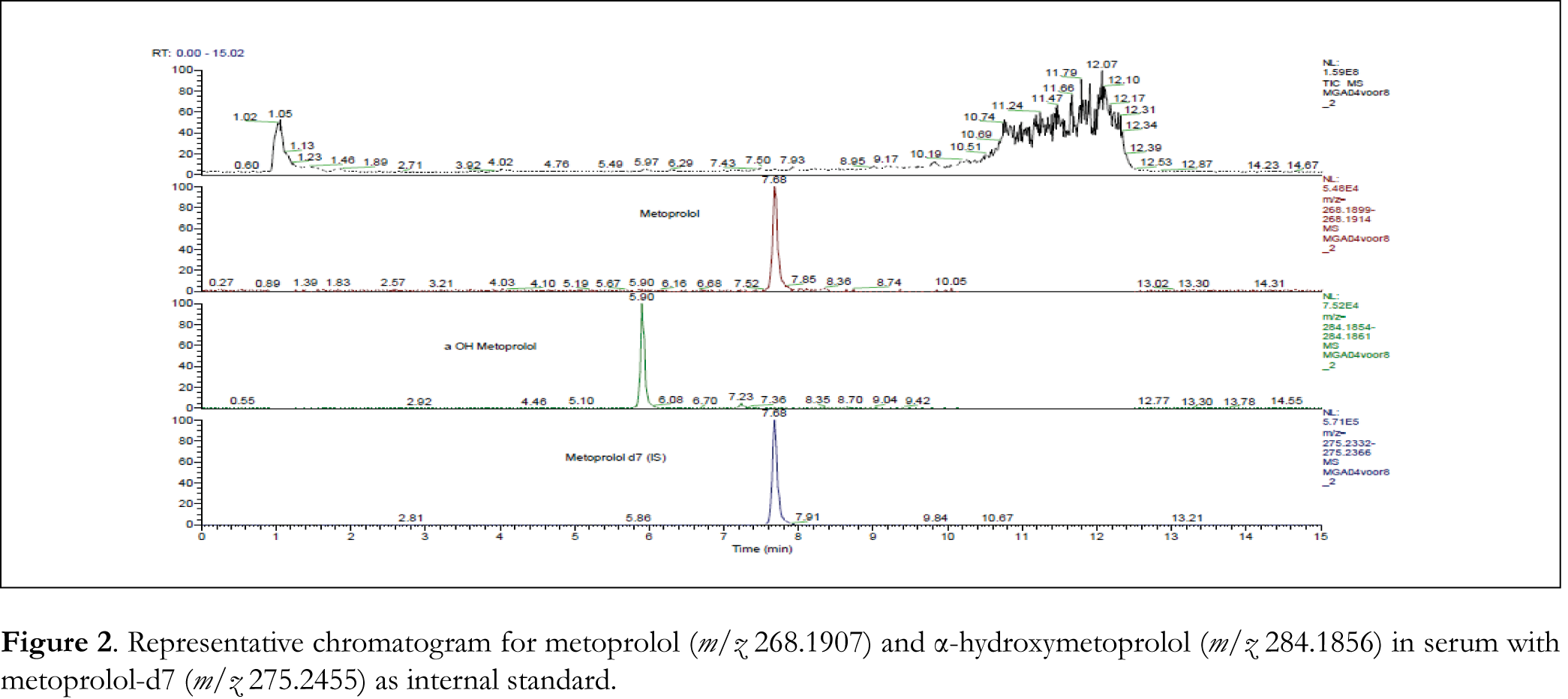
With a total run time of 15.0 min, metoprolol, α-hydroxymetoprolol and IS were exhibited at retention times of 7.7, 5.9 and 7.7 min respectively. Using water:acetonitrile (15-85, v/v) as a solvent for the precipitation of proteins in the sample processing, clean chromatograms were obtained with no significant matrix effect. Metoprolol, α-hydroxymetoprolol and IS gave protonated (M+H)+ ions at m/z 268.1907, 284.1856 and 275.2455 respectively in full scan spectra.
Selectivity
The selectivity of the method was investigated by comparing the chromatograms of blank calf serum, blank human serum and serum from 6 different patients, with known concentration spiked blank calf serum chromatograms. No significant interferences from endogenous substances in serum were found. In Figure 2 a typical chromatogram for metoprolol and α-hydroxymetoprolol in human serum is shown.
Linearity, precision and accuracy
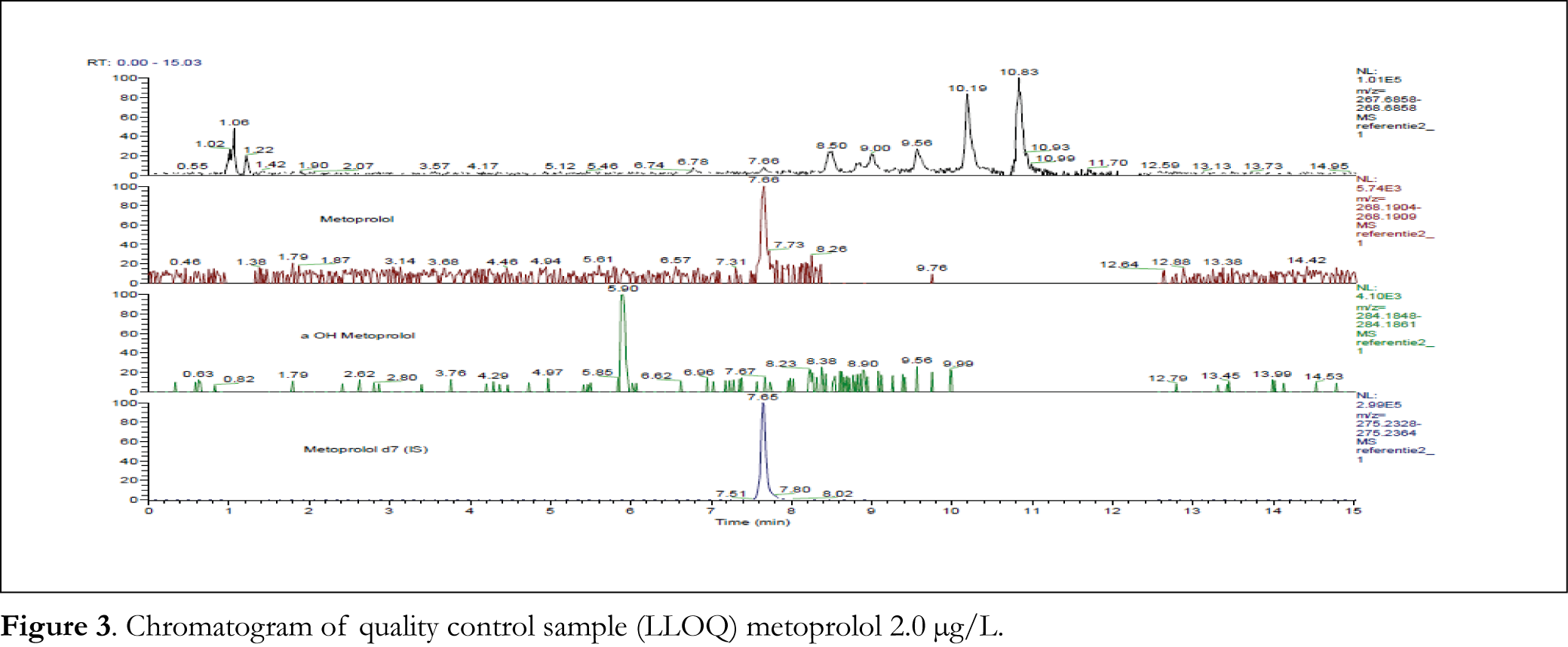
The calibration curve was constructed by plotting the peak area ratios (y) of each analyte to that of the internal standard versus the nominal concentration (x) of each analyte. After comparing the weighting factor models at none, 1/x and 1/x2, the regression equation with weighting factor 1/x for all analytes was found to be the best fit with correlation coefficients (r2) of 0.9990-0.9996 and 0.9963-0.9991 for metoprolol and α-hydroxymetoprolol, respectively. The representative regression equations for calibration curves during the validation were y = 0.01052 x + 0.01728 for metoprolol, and y = 0.00756 x – 0.005723 for α-hydroxymetoprolol. Calibration curves were linear from 5.00 to 250.0 µg/L for both metoprolol and α-hydroxymetoprolol. In common analytical methods where background noise is present, the limits of detection and quantitation are estimated by evaluation of S/N ratios. However, it is difficult to apply this concept to high-resolution accurate mass data when no noise is observed [18]. Rather than defining the LLOQ based on S/N, we determined the LLOQ by the analysis of samples with known concentrations and by establishing the minimum level at which the analytes can be quantified with acceptable accuracy and precision. The LLOQ was found to be 2.0 µg/L for metoprolol (see Figure 3 for chromatogram) and 1.0 µg/L for α-hydroxymetoprolol. Table 1 summarizes the intra- and interday precision and accuracy for metoprolol LLOQ (2.0 µg/L) and QC samples at 5.2, 10.4, 130.6 and 261.1 µg/L, and for α-hydroxymetoprolol LLOQ (1.0 µg/L) and QC samples at 5.0, 10.0, 125.0 and 250.0 µg/L concentrations in calf serum. Intra- and interday precision (RSD) were measured to be within 0.9-7.8% for metoprolol, and within 2.8-10.1% for α-hydroxymetoprolol. Accuracy (RE) were -3.8-5.6% for metoprolol and -0.7-8.3% for α-hydroxymetoprolol. As in pharmacokinetic studies concentrations of metoprolol and α-hydroxymetoprolol were not to be expected higher than the calibration range, dilution integrity was not assessed.
Recovery and matrix effect
Recoveries from QCs in surrogate matrix (blank calf serum) or pooled human serum were 97.7 (RSD 2.4%) and 98.3 (RSD 2.6%) for metoprolol, and 103.0 (RSD 4.1%) and 103.0 (RSD 4.4%) for α-hydroxymetoprolol, respectively. No significant differences (p<0.05) on recoveries were seen between surrogate matrix blank calf serum and blank human serum.
The ionization can be suppressed and enhanced by co-eluting matrix components. Therefore the potential variable matrix related ion suppression was evaluated in 6 independent sources of human serum by calculating the IS normalized matrix factor. For metoprolol the IS normalized matrix factor ranged between 1.04 and 1.26 with an RSD of <4%; for α-hydroxymetoprolol the IS normalized matrix factor ranged between 0.97 and 1.15 with an RSD of <7% (Table 2).
Stabilities
In Table 3 the results from all the stability experiments are shown. Metoprolol and α-hydroxymetoprolol in quality control samples were stable for 24 h at room temperature in daylight as well as in the dark. No significant degradation was observed after 2 freeze-thaw cycles and during autosampler storage for 30 h. Metoprolol and α-hydroxymetoprolol in quality control samples were at least stable at -24 °C for 267 days. All results were within 15% of the nominal concentrations, showing sufficient stability for sample analysis and storage.
Method application
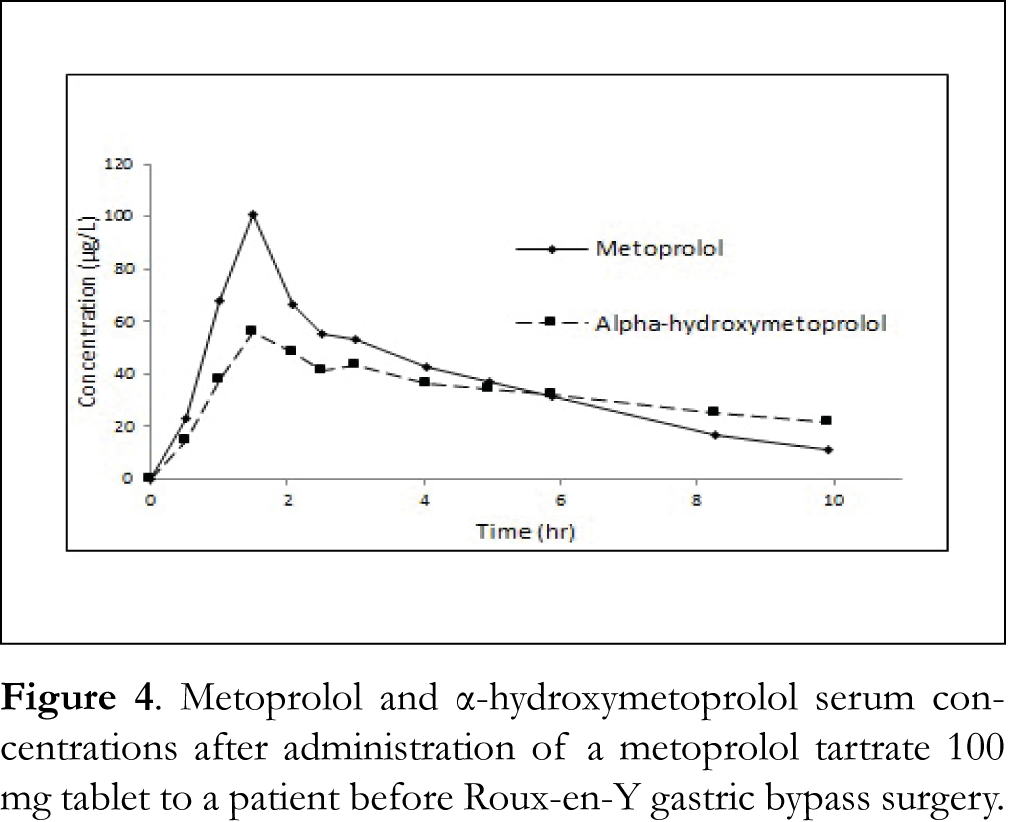
This method will be applied in clinical studies on the influence of Roux-en-Y gastric bypass surgery on the rate and extent of absorption of metoprolol from different oral formulations (Dutch Trial Register numbers NTR4000 and NTR4001). Results will be available shortly. Figure 4 shows serum concentrations of metoprolol and α-hydroxymetoprolol after administration of a metoprolol tartrate 100 mg tablet to a patient before surgery.
Conclusion
This validated LC-Orbitrap MS method for the simultaneous determination of metoprolol and α-hydroxymetoprolol in human serum was simple, selective, precise, accurate and reproducible. Absence of matrix interferences effects was adequately demonstrated. This method can be used for application in human pharmacokinetics.
References
1. Plosker GL, Clissold SP. Controlled release metoprolol formulations: a review of their pharmacodynamic and pharmacokinetic properties, and therapeutic use in hypertension and ischaemic heart disease. Drugs 43, 382-414 (1992). [CrossRef]
2. Aqil M, Ali A, Ahad A, et al. A validated HPLC method for estimation of metoprolol in human plasma. Acta Chromatogr 17, 130-140 (2007).
3. Baranowska I, Magiera S, Baranowski J. UHPLC method for the simultaneous determination of β-blockers, isoflavones and their metabolites in human urine. J Chromatogr B 879, 615-626 (2011). [CrossRef]
4. Mistry B, Leslie J, Eddington NE. A sensitive assay of metoprolol and its major metabolite α-hydroxy metoprolol in human plasma and determination of dextromethorphan and its metabolite dextrorphan in urine with high performance liquid chromatography and fluorometric detection. J Pharm Biomed Anal 16, 1041-1049 (1998). [CrossRef]
5. Albers S, Elshof JP, Völker C, et al. HPLC quantification of metoprolol with solid-phase extraction for the drug monitoring of pediatric patients. Biomed Chromatogr 19, 202-207 (2005). [CrossRef]
6. Yilmaz B, Asci A, Arslan S. Determination of metoprolol in human plasma and urine by high-performance liquid chromatography with fluorescence detection. J Sep Sci 33, 1904-1908 (2010). [CrossRef]
7. Xu T, Bao S, Geng P, et al. Determination of metoprolol and its two metabolites in human plasma and urine by high performance liquid chromatography with fluorescence detection and its application in pharmacokinetics. J Chromatogr B 937, 60-66 (2013). [CrossRef]
8. Yilmaz B, Arslan S, Akba V. Gas chromatography-mass spectrometry method for determination of emtoprolol in the patients with hypertension. Talanta 80, 346-351 (2009). [CrossRef]
9. Senthamil Selvan P, Pal TK. Chromatography-tandem mass spectrometry method for the simultaneous quantitation of metoprolol succinate and simvastatin in human plasma. J Pharm Biomed Anal 49, 780-785 (2009). [CrossRef]
10. Venkateswarlu P, Kumar BN, Seshaiah K, et al. Selective and sensitive method for the determination of metoprolol in human plasma using liquid chromatography coupled with tandem mass spectrometry. Acta Pharm 60, 177-184 (2010). [CrossRef]
11. Bae SH, Lee JK, Cho DY, et al. Simultaneous determination of metoprolol and its metabolites, α-hydroxymetoprolol and O-desmethylmetoprolol, in human plasma by liquid chromatography with tandem mass spectrometry: application to the pharmacokinetics of metoprolol associated with CYP2D6 genotypes. J Sep Sci 37, 1256-1264 (2014). [CrossRef]
12. Antunes NJ, Cavalli RC, Marques MP, et al. Stereoselective determination of metoprolol and its metabolite α-hydroxymetoprolol in plasma by LC MS/MS: application to pharmacokinetics during pregnancy. Chirality 25, 1-7 (2013). [CrossRef]
13. Rao Z, Ma YR, Qin HY, et al. Development of a LC-MS/MS method for simultaneous determination of metoprolol and its metabolites, α-hydroxymetoprolol and O-desmethylmetoprolol, in rat plasma: application to the herb-drug interaction study of metoprolol and breviscapine. Biomed Chromatogr 29, 1453-1460 (2015). [CrossRef]
14. Baranowska I, Adolf W, Magiera S. Enantioselective determination of metoprolol and its metabolites in human urine high-performance liquid chromatography with fluorescence detection (HPLC-FLD) and tandem mass spectrometry (MS/MS). J Chromatogr B 1004, 79-84 (2015). [CrossRef]
15. Sun W, Chen R, Li W, et al. Simultaneous determination of ivabradine, metoprolol and their metabolites in rat plasma by ultra-performance liquid chromatography tandem mass spectrometry and its application in a pharmacokinetic study. Anal Methods 7, 8459-8465 (2015). [CrossRef]
16. Guideline on bioanalytical method validation. European Medicines Agency, Committee for Medicinal products for Human Use. London (2011).
17. Lamiable D, Hoizey G, Oget O, et al. An ultra-high resolution accurate mass LC/MS solution to forensic toxicology screening in serum. Poster (2009) https://tools.thermofisher.com/content/sfs/posters/POS_IMSC_2009Exactive_Lamiable(1).pdf
18. Sturm RM, Jones BR, Mulvana DE, et al. HRMS using Q-Exactive series mass spectrometer for regulated quantitative bioanalysis: how, when, and why to implement. Bioanalysis 8, 1709-1721 (2016). [CrossRef]
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License
