OPEN-ACCESS PEER-REVIEWED
ORIGINAL RESEARCH
Yifan Shi1,*, Heather E. Murrey2, Kay Ahn2, Naidong Weng1, Shefali Patel1
1DMPK, Discovery, Product Development & Supply, Janssen R&D, 2Lead Discovery, Discovery, Product Development & Supply, Janssen R&D, Springhouse, PA, USA.
Journal of Applied Bioanalysis. Vol.6. No.3. pages 131-144 (2020).
Published 15 August 2020. https://doi.org/10.17145/jab.20.014 | (ISSN 2405-710X).
Correspondence: Shi Y. . Janssen R&D, 1400 McKean Road, Spring House, PA 19477, USA. Phone: +1 215 628 6854.
Citation:
Shi Y, Murrey HE, Ahn K, Weng N, Patel S. LC-MS/MS assay for the simultaneous quantitation of thromboxane B2 and prostaglandin E2 to evaluate cyclooxygenase inhibition in human whole blood. J Appl Bioanal 6(3), 131-144 (2020).
Open-access and Copyright:
©2020 Shi Y et al. This article is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Funding/Manuscript writing assistance:
The authors have no financial support or funding to report and they also declare that no writing assistance was utilized in the production of this article.
Competing interest:
The authors have declared that no competing interest exist.
Article history:
Received: 01 May 2020, Revised 14 June 2020, Accepted 16 June 2020.
Abstract
OBJECTIVES: A high-throughput LC-MS method for TXB2 and PGE2 was developed for human whole blood assay for COX inhibition.
METHODS: A surrogate analyte approach was used for the quantitation of TXB2 and PGE2 by LC-MS. Fifty microliters plasma was processed using solid-phase extraction. TXB2-d4 and PGE2-d4 were used as surrogate analytes. The calibration curves were established for TXB2 from 0.1 to 500 ng/mL and for PGE2 from 0.05 to 500 ng/mL. TXB1 was used as internal standard.
RESULTS: The response factor and parallelism between surrogate and authentic analyte were verified. Heparinized whole blood assay for COX inhibition was optimized for sample pretreatment, stimulant concentration and incubation time.
CONCLUSION: The LC-MS assay was successfully used to analyze inhibitory activity of four commercially available COX inhibitors. The presented method offers a sensitive, high throughput and low-cost alternative to ELISA for human whole blood assay for COX inhibition.
Keywords
COX inhibitor, LC-MS/MS, lipids, TXB2, PGE2, whole blood assay
Introduction
Prostaglandin-endoperoxide synthases, commonly known as cyclooxygenase (COX), are responsible for the formation of thromboxanes and prostaglandins from arachidonic acid [1,2]. COX enzymes exist in two isoforms, COX-1 and COX-2. The former is expressed constitutively in many tissues, while the latter is generally induced in cells in response to inflammatory cytokines, growth factors and toxins. Inhibition of COX enzymes is widely studied for treatment of inflammation, pain relief, and cancer. Traditional non-steroidal anti-inflammatory drugs (NSAIDs) show an increased risk of gastrointestinal complications, likely due to inhibition of COX-1. Selective inhibitors of COX-2 were subsequently developed. However, several of them were withdrawn from market due to associated cardiovascular risk [3,4]. Two decades later, the therapeutic and adverse effects of both selective and non-selective NSAIDs are still being widely studied [5,6]. It has been shown that the differences in pharmacokinetic and pharmacodynamic characteristics, as well as the dosage level, are more important in evaluating individual risk and benefit of a drug candidate than simply looking at class effects [3].
Various in vitro assays can be used to characterize a compound’s inhibitory activity and selectivity for COX-1 and/or COX-2. These assays have been developed using purified recombinant enzymes, enzyme expression in cultured cells, or enzyme present from human whole blood [7,8]. Enzyme assays using purified protein can reveal important kinetic parameters of inhibition mechanism. However, they cannot account for the effects of plasma protein binding, cellular permeability, and other interactions between an inhibitor and the complex biomolecules within a cell. While cell-based assays account for some of these parameters, it still does not resemble the physiological condition as many blood components are neglected. Therefore, assays developed to examine inhibitor activity in human whole blood, which closely mimics the in vivo environment, is the ideal system to evaluate compounds of interest in developing new COX inhibitors.
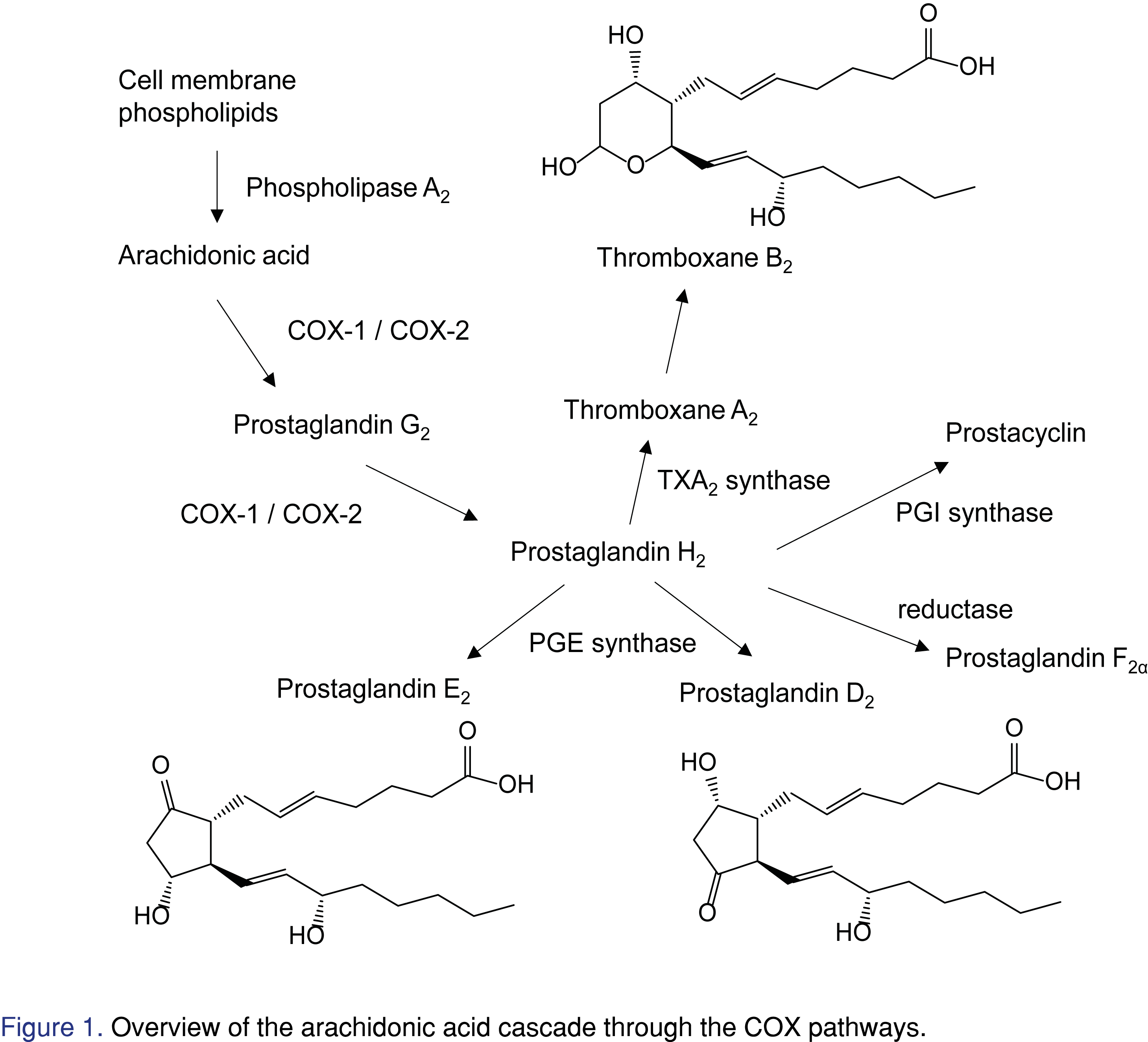
In the COX enzymatic pathway, arachidonic acid is released from cell membrane phospholipids by phospholipase A2. As shown in Figure 1, cyclooxygenase enzymes metabolize arachidonic acid to prostaglandin H2, which can be further converted to thromboxanes and prostaglandins. It has been shown that adding calcium ionophore can stimulate release of arachidonic acid from cellular membrane in vitro, leading COX-1 enzyme to generate thromboxane A2 (TXA2) which is immediately converted to thromboxane B2 (TXB2). Also, lipopolysaccharide (LPS) can induce COX-2 activity, where prostaglandin E2 (PGE2) level is elevated [8]. Therefore, TXB2 and PGE2 levels correlate to COX-1 and COX-2 activities stimulated by calcium ionophore and LPS, respectively. In the human whole blood assay, various concentrations of COX inhibitors are added after COX-1 or COX-2 stimulation in vitro. Plasma samples are collected from the experiment. Inhibition curves of COX-1 and COX-2 activity can be plotted based on the concentrations of TXB2 and PGE2 production to calculate the corresponding half maximal inhibitory concentrations (IC50). Thus, sensitive and high throughput assays for TXB2 (COX-1 activity) and PGE2 (COX-2 activity) in plasma are needed. Commercial enzyme-linked immunosorbent assay (ELISA) kits are available for both compounds. However, plasma samples from whole blood assay need to be analyzed separately using different ELISA kits for PGE2 and TXB2. While the sensitivity of PGE2 and TXB2 detection by the kits is good, the calibration ranges are relatively narrow (from 0.015 to 2 ng/mL). Furthermore, the binding antibodies suffer cross reactivity with other analytes [9]. Multiple LC-MS/MS methods can be found in the literature for detection of PGE2 and related compounds. Excellent sensitivity below 10 pg/mL has been achieved by utilizing chemical derivatization [10]. Direct measurement using LC-MS/MS can also have good sensitivity. However, some methods only cover limited analytes including prostaglandins but not thromboxanes [11], and some methods chromatographically separated a wide range of eicosanoids but require a relatively long run time [12]. Recently, Gandhi et al. utilized a quick protein crash method to analyze five lipids for tissue samples but not in plasma samples [13]. To our best knowledge, this is the first work to report a sensitive and high-throughput LC-MS/MS method for the simultaneous measurement of TXB2 and PGE2 for human whole blood assay.
In the literature, whole blood assays for measurement of COX-2 activity are usually similar to the experiment reported by Patrignani et al. [14]. However, for COX-1 whole blood assays, there are three different procedures, including platelet assays, blood clotting assays, and heparinized whole blood assays. While the platelets isolated from freshly collected whole blood represent most of the COX-1 enzyme activity in blood, platelet assays are not true whole blood assays. Young et al. developed a heparinized blood assay that measures COX-1 and COX-2 activities from the same blood samples by monitoring TXB2 [15]. Esser et al. simplified the heparinized blood procedure by splitting samples into COX-1 and COX-2 inhibition after incubation with COX inhibitors [16]. These experiments resemble physiological conditions and have less individual variation as the same blood samples were used for both COX-1 and COX-2 assays. The measured COX inhibition and selectivity are believed to be more relevant in drug design and development. In this work, we further optimized the heparinized whole blood procedure for both COX-1 and COX-2 assays. Several non-selective and selective NSAIDs were evaluated for their COX-1 and COX-2 inhibitory activities in human whole blood. The calculated IC50 from LC-MS/MS data were compared with those calculated from ELISA analysis.
Experimental
Chemicals and Reagents
PGE2, PGD2, TXB2, PGE2-d4, TXB2-d4, and TXB1 were purchased from Cayman Chemical (Ann Arbor, MI, USA). Methanol and acetonitrile of HPLC grade were obtained from EMD Millipore (Billerica, MA, USA). Formic acid (reagent grade), LPS from E. coli, calcium ionophore, celecoxib, rofecoxib, etoricoxib, and diclofenac were purchased from Sigma-Aldrich (St. Louis, MD, USA). Deionized water was purified via Milli-Q system from EMD Millipore, and cell culture grade water was from Mediatech, Inc (Manassas, VA, USA). TXB2 and PGE2 Express ELISA kits, which contained analyte standards, antibodies, tracer, coated plates, Ellman’s reagent, wash and dilution buffers, were purchased from Cayman Chemical. Human blood from donors who had received no NSAIDs for at least 10 days was collected into vacutainers containing heparin and used within 1 hour of collection.
Instrumentation
For the LC-MS/MS method, all analyses were conducted on a Triple Quad 6500+ mass spectrometer from Sciex (Foster City, CA, USA) with a Turbo Ionspray interface. The system was controlled by Analyst 1.6 software. The LC system used was a Shimadzu LC-30AD pump coupled with a Nexera X2 SIL-30AC autosampler (Kyoto, Japan). Plasma samples were extracted using Oasis MAX 96-well μElution plate from Waters (Milford, MA). A Kinetex C18 column (2 x 50 mm, 2.6 μm) from Phenomenex (Torrance, CA, USA) was used for chromatographic separation. For the ELISA method, the plate reader used was a SpectraMax 250 from Molecular Devices (San Jose, CA, USA) controlled by SoftMax Pro 5.4.5 software
LC-MS/MS and ELISA Methods
To simultaneously measure TXB2 and PGE2 using LC-MS/MS, 50 μL of plasma sample was mixed with 20 μL of internal standard solution (200 ng/mL TXB1 in water/methanol 50:50), and 500 μL water. An Oasis MAX μElution plate was conditioned with 200 μL of methanol and 200 μL of acetonitrile/water 25:75. After vortex mixing, the samples were loaded to SPE plate and washed with 200 μL of acetonitrile/water 25:75 and then 200 μL of freshly prepared 5% ammonia in water. The plate was dried with positive pressure for 1 minute. Analytes were eluted with 40 μL of 1% formic acid in acetonitrile/water 50:50 and 40 μL of 1% formic acid in acetonitrile. The samples were vortex-mixed well and centrifuged for 1 minute. Five microliters sample extract was injected onto the LC-MS/MS system for analysis.
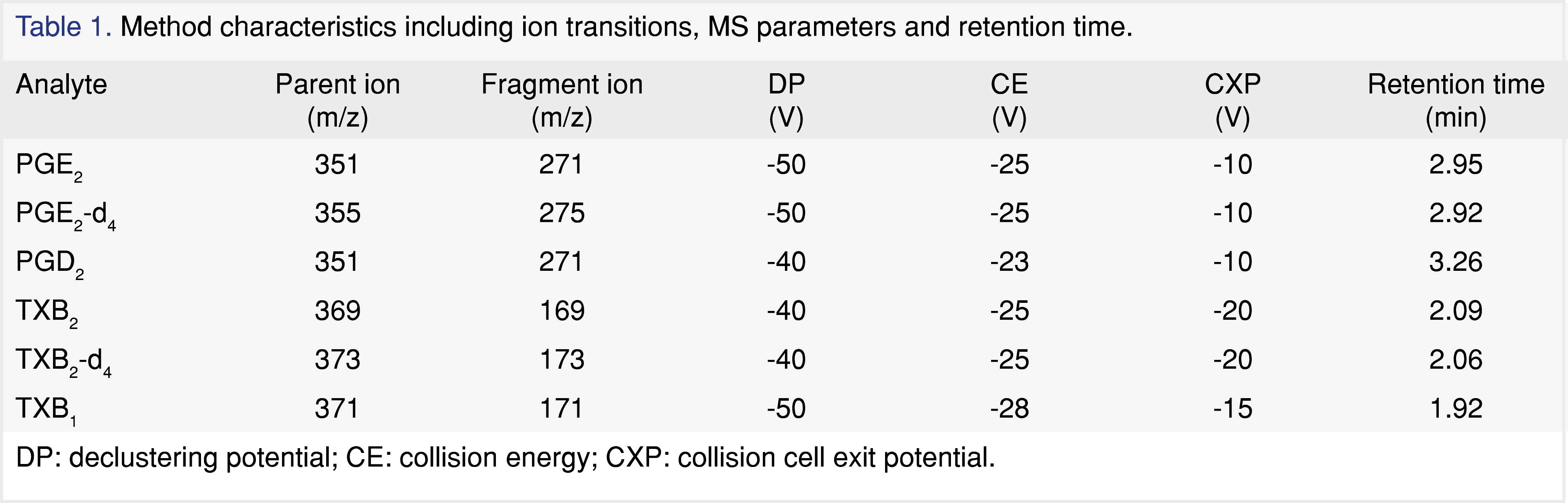
The Kinetex C18 column was maintained at ambient temperature. The mobile phase A was 0.1% acetic acid in water, and the mobile phase B was acetonitrile/isopropanol 90:10. The gradient at an initial flow rate of 0.4 mL/min was ramped from 30% B to 35% B between 0 and 3.2 min. From 3.3 to 3.9 min, the column was forward washed at 0.6 mL/min and 90% B. After the flow rate was dropped back to 0.4 mL/min, the column was re-equilibrated at 30% from 3.91 to 4.4 min. Mass spectrometric detection using negative electrospray ionization (ESI) was carried out in selected reaction monitoring (SRM) mode. The ion spray voltage was -4500 V. The ion source temperature was 600 °C and the entrance potential (EP) was -10 V. Curtain gas, gas 1 and gas 2 were 30, 70 and 35 psi. Collision-activated dissociation (CAD) gas was set to 10 psi. For the transition of each analyte, declustering potential (DP), collision energy (CE), and collision cell exit potential (CXP) were optimized as listed in Table 1. A weighted linear 1/x2 regression was used to generate the calibration curve. Data analysis was carried out with Sciex MultiQuant 2.1.1.
For the ELISA method, plasma samples were analyzed for TXB2 and PGE2 using TXB2 Express and PGE2 Express ELISA kits, respectively, according to the manual. All calibration standards (15–2000 pg/mL) and samples were prepared in duplicates. Sample concentrations were calculated using a four-parameter fit calibration curve with SoftMax Pro 5.4.5.
Figures and Tables


[Click to enlarge]
Method qualification and parallelism assessment
Method qualification and stability
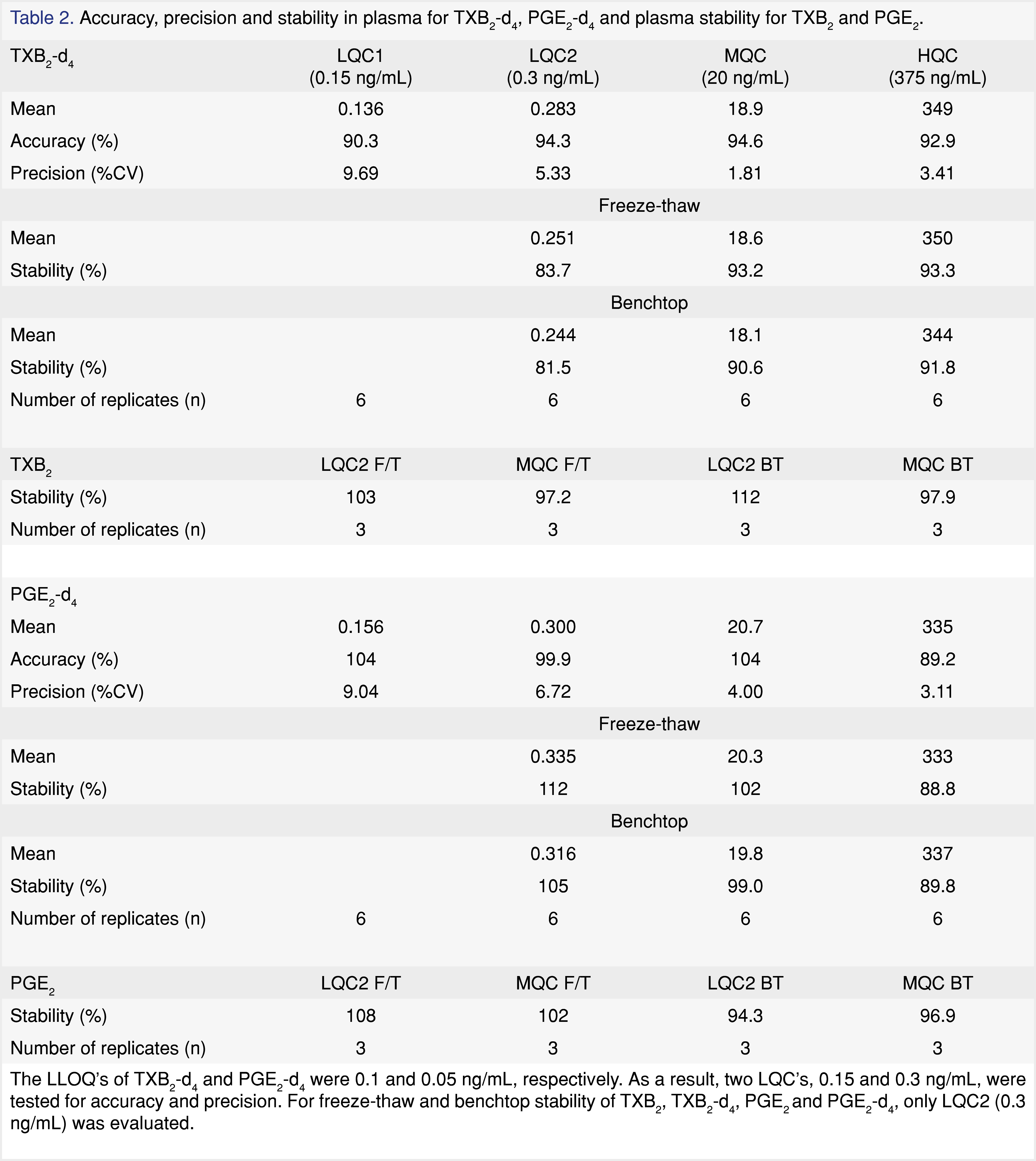
The method qualification was carried out in accordance to internal bioanalytical guideline. The qualification batch contained a calibration curve, six replicates of QCs, benchtop and freeze-thaw stability QCs. The linearity of the calibration curve was determined using a linear 1/x2 weighted regression model. Calibration standards in human plasma were prepared fresh daily with concentration of 0.05, 0.1, 0.5, 2.5, 10, 50, 250, 400, and 500 ng/mL. The lower limit of quantitation (LLOQ) of TXB2 and PGE2 were determined to be 0.1 and 0.05 ng/mL (S/N ratio ≥ 5:1), respectively. Four levels of quality control (QC) samples were prepared at concentrations of 0.15, 0.3, 20, and 375 ng/mL. The intra-day accuracy and precision were assessed by analyzing six replicates of QC samples per level. The interday assay precision was not evaluated for method qualification. Assay accuracy and precision acceptance criteria were within 100 ± 20% according to internal bioanalytical guideline. Standards and QCs were prepared using surrogate analyte (TXB2-d4 and PGE2-d4) in authentic matrix. Stability of the surrogate (d4) and the authentic analyte (d0) in human plasma were assessed for three freeze-thaw cycles and four hours at room temperature. A percent deviation from of ± 20% was deemed acceptable to consider the sample stable under the storage condition tested.
Response Factor and parallelism
For this surrogate analyte method, the mass spectrometry response of the surrogate analyte should be comparable to that of the authentic analyte. The response factor was determined by separately injecting neat solutions of authentic and surrogate analyte in the presence of the internal standard. If the mean percent difference is greater than 5%, the response factor should be applied for quantitation using the surrogate analyte standard curve. A parallelism assessment was performed to determine how well calibration standards prepared with the surrogate analyte track the response of the authentic analyte in the biological matrix [17]. Six replicates of plasma spiked with solvent-only or with authentic analyte at three concentrations (0.3, 20 and 375 ng/mL) were quantified using surrogate analyte curve. Theoretical concentrations of the spiked QC were calculated from the endogenous level plus the spiked amount. The precision and accuracy of the QCs were evaluated to establish parallelism across the calibration range [18].
COX-1 and COX-2 whole blood assay
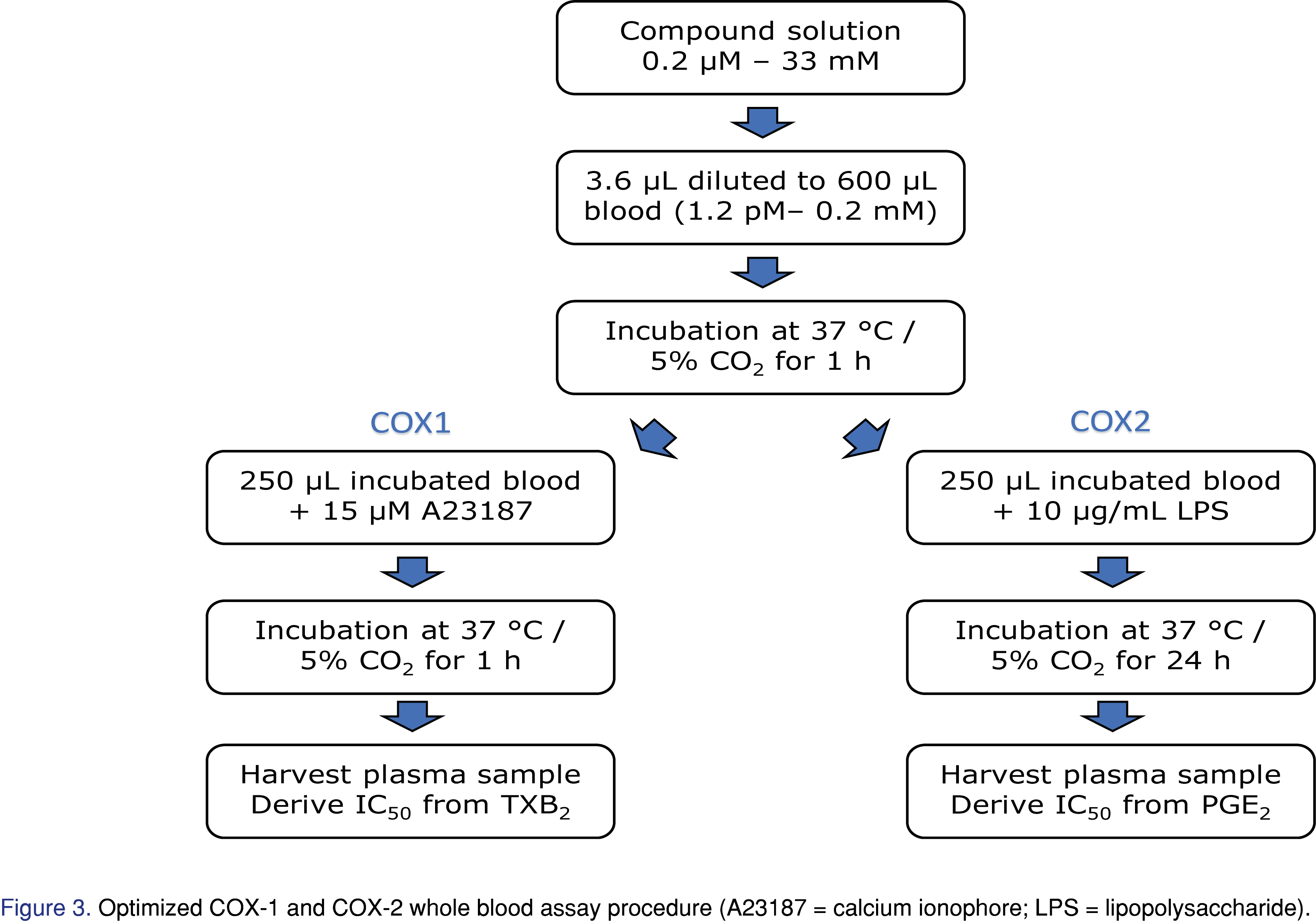
Fresh blood was collected in heparinized vacutainers from volunteers with consent. Protocol for Employee Volunteer Participation in Sample Donation for Research was under the governance of Quorum Institutional Review Board (reference Pro00035367). The subjects had not taken any NSAIDs for at least 10 days prior to blood collection. The first and highest compound spiking solution was prepared at 33.3 mM in DMSO. Ten additional spiking solutions were subsequently diluted 3.33x from the previous solution. The lowest solution was 0.2 μM. A blank DMSO solution completed the 12-point inhibition curve. The final compound concentrations ranged from 1.2 pM to 0.2 mM (167,000 folds). COX inhibitors were added within one hour of blood collection. All compounds, celecoxib, rofecoxib, diclofenac and etoricoxib, were tested in duplicates. The samples were pipetted mixed 10 times prior to incubation in a humidified 37 °C incubator with 5% carbon dioxide for 1 hour. The samples were then divided equally into two aliquots for COX-1 and COX-2 experiments.
For COX-1, 2 μL of calcium ionophore in DMSO was added to each well resulting in a final concentration of 15 μM. After pipette mixing of the samples, the plate was incubated at 37 °C for one hour with continuous shaking at 200 rpm. The reactions were terminated by quickly chilling the plates in an ice batch, centrifuging at 1100 x g for 10 minutes to separate the plasma and storing the separated plasma at −80 °C. For COX-2, 10 μL of LPS stock solution in H2O was added to blood samples resulting a final concentration of 10 μg/mL. After mixing, the plate was incubated at 37 °C with 5% carbon dioxide for 24 hours. Plasma were collected by centrifuging at 1100 x g for 10 minutes.
Plasma samples from the whole blood assay were analyzed with both ELISA and LC-MS/MS methods. TXB2 and PGE2 concentration data were analyzed using Prism 7.0 (GraphPad Software, San Diego, CA) and fitted to a four-parameter logistic function using nonlinear regression analysis. IC50 values of COX-1 and COX-2 inhibition were calculated based on the inhibition curve.
Results and Discussion
LC-MS/MS method development and qualification
Two strategies were commonly used for analyzing endogenous compound concentration in biological matrix: surrogate analyte in authentic matrix and authentic analyte in surrogate matrix [19]. We used the first approach in which the mass spectrometry response of authentic and surrogate analyte must be investigated. Six replicates of TXB2 and PGE2 neat solution at 5 ng/mL were injected into LC-MS/MS system with the presence of internal standard TXB1. The analyte-to-IS peak area ratio were compared to that of TXB2-d4 and PGE2-d4. The mean response factor was 0.99. Therefore, the response factor was not applied during sample analysis.
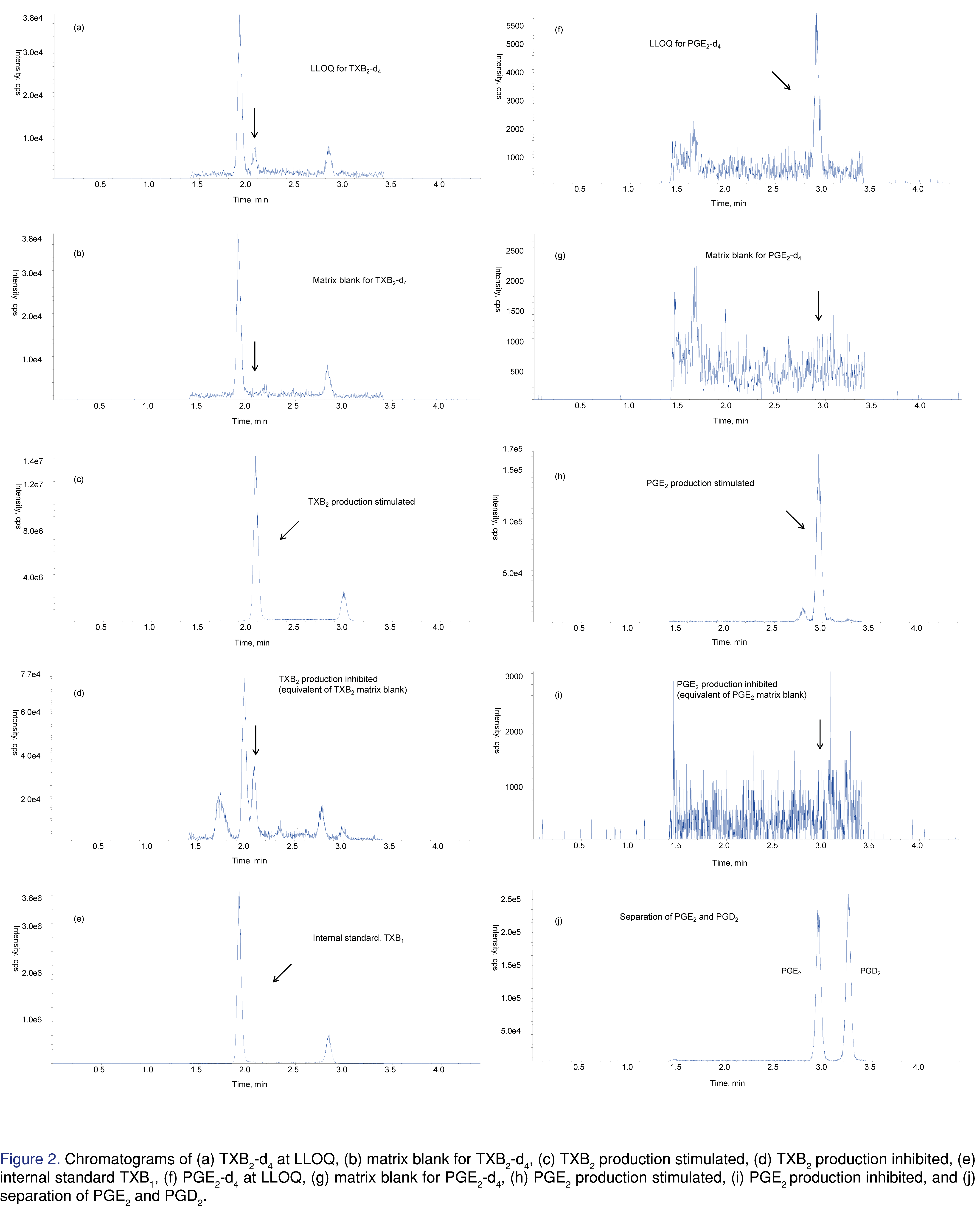
The chromatograms of the surrogate and authentic analytes as well as the internal standard are shown in Figure 2. While mobile phases with formic acid or acetic acid were used in previous publications for TXB2 and PGE2, 0.1% acetic acid was found to achieve the best sensitivity [20]. Different reversed phase columns were tested. The peak shape of PGE2 was generally good on most columns. However, interference peaks were observed at a slightly earlier retention time for TXB2 and TXB2-d4. Kinetex C18 column provided the best separation of the interference peak. Gandhi et al. reported that TXB2 frequently had peak tailing at elevated column temperature (40 °C), so the column temperature should be kept at 20 ~ 25 °C [13]. We found that adding 10% isopropanol to mobile phase B also reduced the peak tailing for TXB2. No interference peak was observed for the internal standard, TXB1. Prostaglandin D2 and E2 are isobaric compounds and must be chromatographically resolved. Using an analytical flow of 0.4 mL/min and a column-wash flow of 0.6 min/mL, we achieved baseline separation using only 4.4 minutes of HPLC time. As shown in Figure 2 (i), PGD2 and PGE2 were eluted at 2.95 min and 3.26 min, respectively. To establish parallelism of the surrogate and authentic analytes in human plasma, endogenous levels of TXB2 and PGE2 were determined using surrogate analyte curve. The measured TXB2 and PGE2 concentrations using 6 replicates of blank human plasma were 0.701 and 0.339 ng/mL, respectively. The same lot of human plasma was used to make three levels of QCs by spiking in authentic analyte solution at 0.3, 20 and 375 ng/mL. The addition of these QCs was determined by the same surrogate analyte curve. The relative error of these QC was within 10.3% for both TXB2 and PGE2.
The LC-MS/MS method was qualified in one accuracy and precision batch with matrix stability test. Calibration curves were regressed by plotting the analyte-to-IS peak area ratio versus the concentration. Linear with 1/x2 weighing was determined to be the best curve fit for the range of 0.1 – 500 ng/mL for TXB2-d4 and 0.05 – 500 ng/mL for PGE2-d4. The correlation coefficients for both curves were ≥ 0.99. Calibration standards were within 88.6% and 106% of their nominal values for TXB2-d4 and within 82.3% and 114% for PGE2-d4. The assay accuracy was determined by calculating the ratios of the calculated concentrations to their nominal values, and the intraday precision was determined using a one-way analysis of variance (ANOVA). As presented in Table 2, the overall assay accuracies were within 100 ± 9.7% and within 100 ± 10.8% for TXB2-d4 and PGE2-d4, respectively. The precisions were less than 9.69% CV for TXB2-d4 and less than 9.04% CV for PGE2-d4. The freeze-thaw stability was accessed at three QC concentrations. After three cycles, the stability for both TXB2-d4 and PGE2-d4 was above 83.7%. After 4 hours at room temperature, the stability for the surrogate analyte was above 81.5%. The matrix stability for the authentic analytes, TXB2 and PGE2, was tested at 0.3 and 20 ng/mL. Stability samples going through 3 freeze-thaw cycles or 4 hours on benchtop were compared against the samples stored at -80 °C immediately after preparation. Freeze-thaw and benchtop stability for TXB2 were above 97.2% and 97.9%, respectively, and were above 96.9% and 94.3% for PGE2, respectively.
Whole blood assay optimization and application
The design of whole blood assay
For the research of COX inhibitors, in vitro screening assays, utilizing purified enzymes, cell culture and human whole blood, have been developed. While enzyme assay is fast, it only measures the effect of the drug on the enzyme itself. Cell assay can reflect drug-cell interactions, but still neglects plasma protein binding of the drug and other blood compounds. Thus, we are particularly interested in human whole blood assays. COX-1 human blood assay described in the literature falls into three general categories: freshly isolated platelet assay, blood clotting assay, and heparinized whole blood assay. Platelets constitutively express COX-1. Patrono et al. showed that TXB2 production represents platelet COX-1 activity in response to endogenously formed thrombin [21]. Thus, COX-1 potency can be calculated by measuring TXB2 concentration from platelet assay. However, purified platelet does not resemble physiological conditions, so its results can hardly be conferred to the human organism. Patrignani et al. used unheparinized human whole blood and measured TXB2 production for COX-1 inhibition after spontaneous blood clotting [14]. Brideau et al. further demonstrated that TXB2 production in spontaneously clotted blood reached a plateau after 60 min. While the TXB2 level was close to the baseline within the first few minutes, at as early as 30 min it got to ~50% of the level at 60 min [7]. As a result, to accurately access the compounds’ COX-1 inhibition, testing compounds at concentrations covering a full inhibition curve should be added to sub-aliquoted blood as soon as the blood was collected. With the set up and blood handling requirement of our on-site clinic and our lab, we had difficulty executing the blood clotting COX-1 assay to achieve proper result. For the third option, Young et al. and Glaser et al. described methods using heparinized human whole blood with calcium ionophore incubation for COX-1 inhibition [15,22]. Esser et al. modified the method by having blood samples for COX-1 and COX-2 incubated together with testing compounds [16]. The advantage of this approach is to have testing compound incubated in whole blood before splitting into COX-1 and COX-2 assays, minimizing the variations between the inhibited samples going into next steps. On the other hand, COX-1 platelet and unheparinized blood assays need to be processed separately from COX-2 assays because the blood used for COX-2 must contain anti-coagulant to allow for the incubation with LPS. We used a slightly modified approach for the heparinized blood assay as demonstrated in Figure 3. The blood samples used for COX-1 were split from those of COX-2 after incubation with the testing compounds, which kept only one set of samples in the process until immediately before stimulants were added.
COX-2 whole blood assays in the literature frequently have a preincubation step with aspirin. Constitutively expressed COX-1 in platelet can also generate PGE2 which can be irreversible acetylated by aspirin. The rapid enzymatic hydrolysis of aspirin by plasma esterases ensured that no intact drug was present in whole blood by the time of LPS stimulation, so the LPS stimulated COX-2 activity is not affected. The final PGE2 readouts reflects COX-2 inhibition of the testing compounds. However, PGE2 production by platelets only accounts for 1~2% of cyclooxygenase products and its relative contribution to LPS stimulated whole blood PGE2 production was marginal even without aspirin exposure in vivo or in vitro [14]. Furthermore, the expression of COX-1 stays steady after incubation with LPS for up to 24 hours. Thus, under the experimental conditions commonly used for COX-2 whole blood assay, it is not necessary to pre-incubate the blood with aspirin to inhibit COX-1 activity [7].
Whole blood assay optimization
After the freshly collected heparinized human blood was incubated with the test compound for an hour, the samples were divided into two aliquots. To determine COX-1 activity, calcium ionophore was added to one of the aliquots. After 1 hour of incubation at 37 °C, plasma was harvested for the measurement of TXB2. Calcium ionophore concentrations ranging from 5 to 25 μM were tested. As shown in Figure 4a, while TXB2 production level increased as the concentration of the stimulant increased, it leveled off and trended lower at 20 and 25 μM. At even higher calcium ionophore concentration, e.g. at 50 μM, various degrees of hemolysis were observed in collected plasma samples. We chose 15 μM calcium ionophore concentration for our experiment. We also tested calcium ionophore incubation time from 15 minutes to 3 hours. As expected, TXB2 was released very rapidly upon platelet activation, and no obvious trend was noted. An incubation time of 1 hour was selected.
The second aliquot of the drug-inhibited blood sample was used to determine COX-2 potency with LPS stimulation. Various concentrations of LPS at 0.05, 0.1, 0.5, 1, 5, 10, 50, and 100 μg/mL were tested for COX-2 stimulation. The resulted PGE2 concentration didn’t show any obvious trend. This was evidenced by the wide range of LPS concentrations in previously reported COX-2 whole blood experiments. We chose 10 μg/mL as the LPS incubation concentration for our experiment. PGE2 productions from various LPS incubation time at 2, 4, 7, 24 and 30 hours, were plotted in Figure 4b. PGE2 concentration increased more than 10 folds from 2 h to 24 h and then dropped down at 30 hours. While we cannot rule out the possibility that the most optimum incubation time occurs at the middle point of our tested time profile. The overall increase from 7 to 24 hours incubation was not significant, so we only investigated time frames in which the experiment could be finished within the same day of blood collection or on the following day. As a result, 24 hours LPS incubation was chosen for our experiment.
Comparison of LC-MS/MS and ELISA results
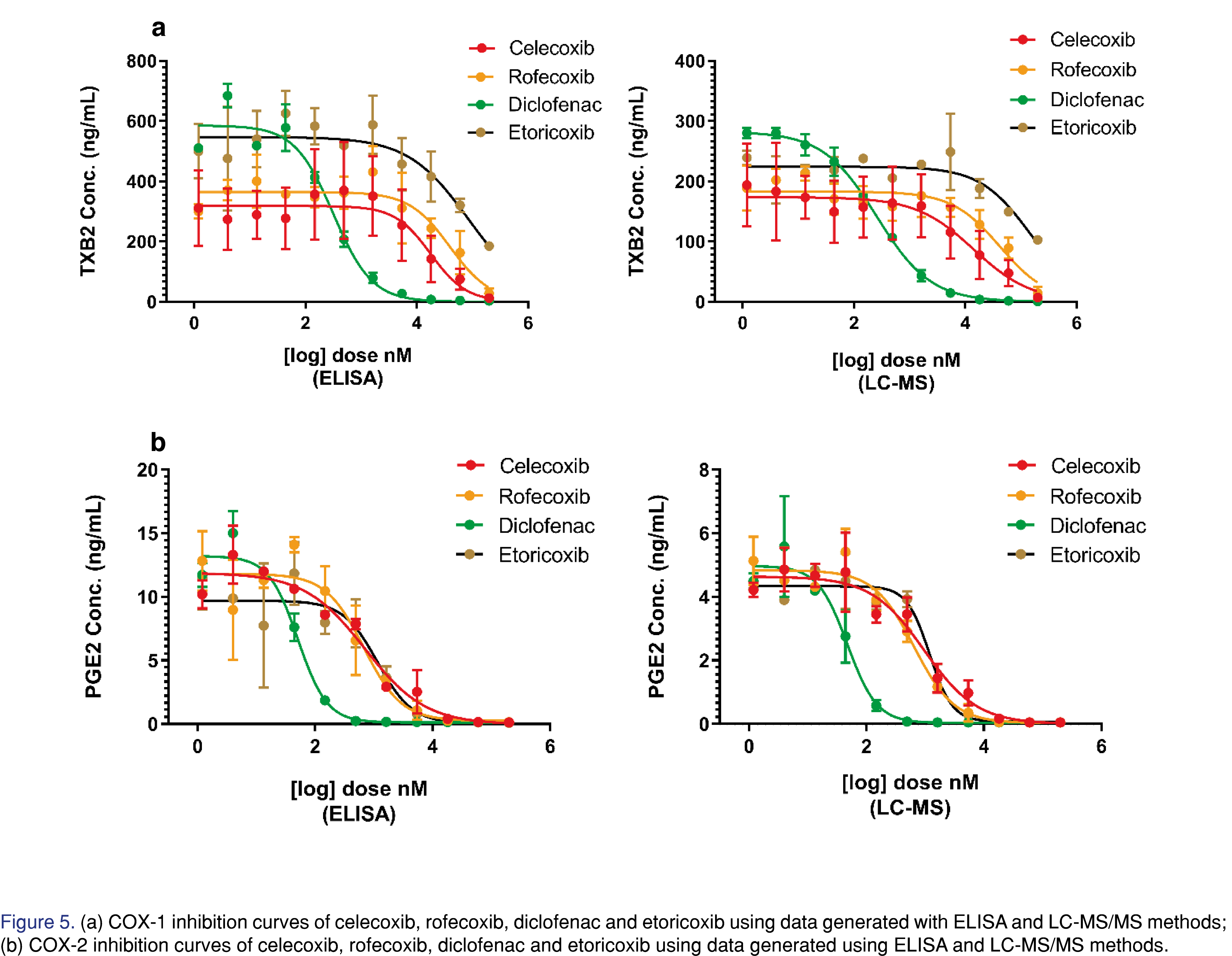
Four COX inhibitors, celecoxib, rofecoxib, diclofenac and etoricoxib, were tested in duplicates using optimized whole blood assay conditions. Plasma samples collected from the whole blood assay were analyzed using both LC-MS/MS and ELISA methods. We developed the sensitive LC-MS/MS method to simultaneously analyze TXB2 and PGE2 in a large linear range. While the instrument running time was longer, the more labor-intensive sample process time was shorter and no over-curve re-assay was needed because of the large curve. Two ELISA kits were used to analyze COX-1 and COX-2 assay samples separately. For TXB2, most of the samples were at or above 100 ng/mL which was significantly higher than the upper limit of quantitation at 2 ng/mL for the ELISA assay. Between the initial analysis and over-the-curve re-assay, four batches were needed for analyzing TXB2 and PGE2 samples using ELISA. With the LC-MS/MS method, all samples were analyzed in one batch.
COX-1 and COX-2 inhibition curves generated by two different analytical methods are shown in Figure 5. While the shapes of the inhibition curve were similar, the absolute levels of the quantitation results were quite different. For both TXB2 and PGE2 ELISA assays, the polyclonal binding antibodies were known to have cross-reactivity with various metabolites, resulting in 100%~200% overestimation compared to the more specific LC-MS/MS method. As shown in Table 3, the calculated IC50 values from both analytical methods were comparable for all four compounds except for that of COX-1 inhibition of etoricoxib. The results from ELISA (85.6 μM) and LC-MS/MS (149 μM) were both close to the upper limit of concentration range (1.2 pM – 200 μM). For highly selective COX-2 inhibitors like etoricoxib, higher compound concentrations are needed to complete its COX-1 inhibition curve at its fully inhibited side (Figure 5a).
Conclusion
In the present study, an LC-MS/MS method for the quantitation of TXB2 and PGE2 for the plasma samples of COX inhibition human whole blood assay was developed using surrogate analyte approach. The LC-MS/MS method eliminates the cross-reactivity with metabolites found in ELISA kits. The assay is sensitive and high-throughput. The response factor and parallelism between surrogate and authentic analyte were investigated. A response factor was not applied when determining analyte concentration from the surrogate analyte calibration curve since the difference was less than 5%. Whole blood COX inhibition assay was optimized for sample pretreatment, stimulant concentration and incubation time. The qualified TXB2/PGE2 combo assay was used to measure the COX-1 and COX-2 IC50 of four COX inhibitors. The IC50 values were comparable between the LC-MS/MS and ELISA methods. In the future, the method can also be used in plasma and tissue samples of animal species, such as rat and mouse, that contain antibodies interfering with commercial ELISA kits.
References
- Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294(5548), 1871-1875 (2001).
https://doi.org/10.1126/science.294.5548.1871 - Buczynski MW, Dumlao DS, Dennis EA. Thematic Review Series: Proteomics. An integrated omics analysis of eicosanoid biology. J Lipid Res 50(6), 1015-1038 (2009).
https://doi.org/10.1194/jlr.R900004-JLR200 - Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest 116(1), 4-15 (2006).
https://doi.org/10.1172/JCI27291 - Abraham NS, El-Serag HB, Hartman C, Richardson P, Deswal A. Cyclooxygenase-2 selectivity of non-steroidal anti-inflammatory drugs and the risk of myocardial infarction and cerebrovascular accident. Aliment Pharmacol Ther 25(8), 913-924 (2007).
https://doi.org/10.1111/j.1365-2036.2007.03292.x - Beales ILP. Time to reappraise the therapeutic place of celecoxib. Ther Adv Chronic Dis 9(5), 107-110 (2018).
https://doi.org/10.1177/2040622317749394 - Barcella CA, Lamberts M, McGettigan P et al. Differences in cardiovascular safety with non-steroidal anti-inflammatory drug therapy-A nationwide study in patients with osteoarthritis. Basic Clin Pharmacol Toxicol 124(5), 629-641 (2019).
https://doi.org/10.1111/bcpt.13182 - Brideau C, Kargman S, Liu S et al. A human whole blood assay for clinical evaluation of biochemical efficacy of cyclooxygenase inhibitors. Inflamm Res 45(2), 68-74 (1996).
https://doi.org/10.1007/BF02265118 - Gierse JK, Zhang Y, Hood WF et al. Valdecoxib: assessment of cyclooxygenase-2 potency and selectivity. J Pharmacol Exp Ther 312(3), 1206-1212 (2005).
https://doi.org/10.1124/jpet.104.076877 - Willenberg I, Meschede AK, Schebb NH. Determining cyclooxygenase-2 activity in three different test systems utilizing online-solid phase extraction-liquid chromatography-mass spectrometry for parallel quantification of prostaglandin E(2), D(2) and thromboxane B(2). J Chromatogr A, 1391 40-48 (2015).
https://doi.org/10.1016/j.chroma.2015.02.059 - Zhu H, Zhuang X, Liu S et al. Ultrahigh-performance liquid chromatography/tandem mass spectrometry method for evaluating enzyme activity and screening inhibitors of cyclooxygenase-2. Rapid Commun Mass Spectrom 28(16), 1792-1800 (2014).
https://doi.org/10.1002/rcm.6963 - Cao H, Yu R, Tao Y, Nikolic D, van Breemen RB. Measurement of cyclooxygenase inhibition using liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal 54(1), 230-235 (2011).
https://doi.org/10.1016/j.jpba.2010.08.001 - Shinde DD, Kim KB, Oh KS et al. LC-MS/MS for the simultaneous analysis of arachidonic acid and 32 related metabolites in human plasma: Basal plasma concentrations and aspirin-induced changes of eicosanoids. J Chromatogr B Analyt Technol Biomed Life Sci 911, 113-121 (2012).
https://doi.org/10.1016/j.jchromb.2012.11.004 - Gandhi AS, Budac D, Khayrullina T, Staal R, Chandrasena G. Quantitative analysis of lipids: a higher-throughput LC-MS/MS-based method and its comparison to ELISA. Future Sci OA 3(1), FSO157 (2017).
https://doi.org/10.4155/fsoa-2016-0067 - Patrignani P, Panara MR, Greco A et al. Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthases. J Pharmacol Exp Ther 271(3), 1705-1712 (1994).
- Young JM, Panah S, Satchawatcharaphong C, Cheung PS. Human whole blood assays for inhibition of prostaglandin G/H synthases-1 and -2 using A23187 and lipopolysaccharide stimulation of thromboxane B2 production. Inflamm Res 45(5), 246-253 (1996).
https://doi.org/10.1007/BF02259611 - Esser R, Berry C, Du Z et al. Preclinical pharmacology of lumiracoxib: a novel selective inhibitor of cyclooxygenase-2. Br J Pharmacol 144(4), 538-550 (2005).
https://doi.org/10.1038/sj.bjp.0706078 - Jones BR, Schultz GA, Eckstein JA, Ackermann BL. Surrogate matrix and surrogate analyte approaches for definitive quantitation of endogenous biomolecules. Bioanalysis 4(19), 2343-2356 (2012).
https://doi.org/10.4155/bio.12.200 - Liu L, Cui Z, Deng Y, Dean B, Hop CE, Liang X. Surrogate analyte approach for quantitation of endogenous NAD(+) in human acidified blood samples using liquid chromatography coupled with electrospray ionization tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 1011, 69-76 (2016).
https://doi.org/10.1016/j.jchromb.2015.12.040 - Jian W, Edom RW, Weng N. Important considerations for quantitation of small-molecule biomarkers using LC-MS. Bioanalysis 4(20), 2431-2434 (2012).
https://doi.org/10.4155/bio.12.247 - Lubin A, Geerinckx S, Bajic S et al. Enhanced performance for the analysis of prostaglandins and thromboxanes by liquid chromatography-tandem mass spectrometry using a new atmospheric pressure ionization source. J Chromatogr A 1440, 260-265 (2016).
https://doi.org/10.1016/j.chroma.2016.02.055 - Patrono C, Ciabattoni G, Patrignani P et al. Clinical pharmacology of platelet cyclooxygenase inhibition. Circulation 72(6), 1177-1184 (1985).
https://doi.org/10.1161/01.CIR.72.6.1177 - Glaser K, Sung ML, O’Neill K et al. Etodolac selectively inhibits human prostaglandin G/H synthase 2 (PGHS-2) versus human PGHS-1. Eur J Pharmacol 281(1), 107-111 (1995).
https://doi.org/10.1016/0014-2999(95)00302-2
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License
